
Running nextflow rnaseq pipeline
Running the Nextflow rnaseq pipeline
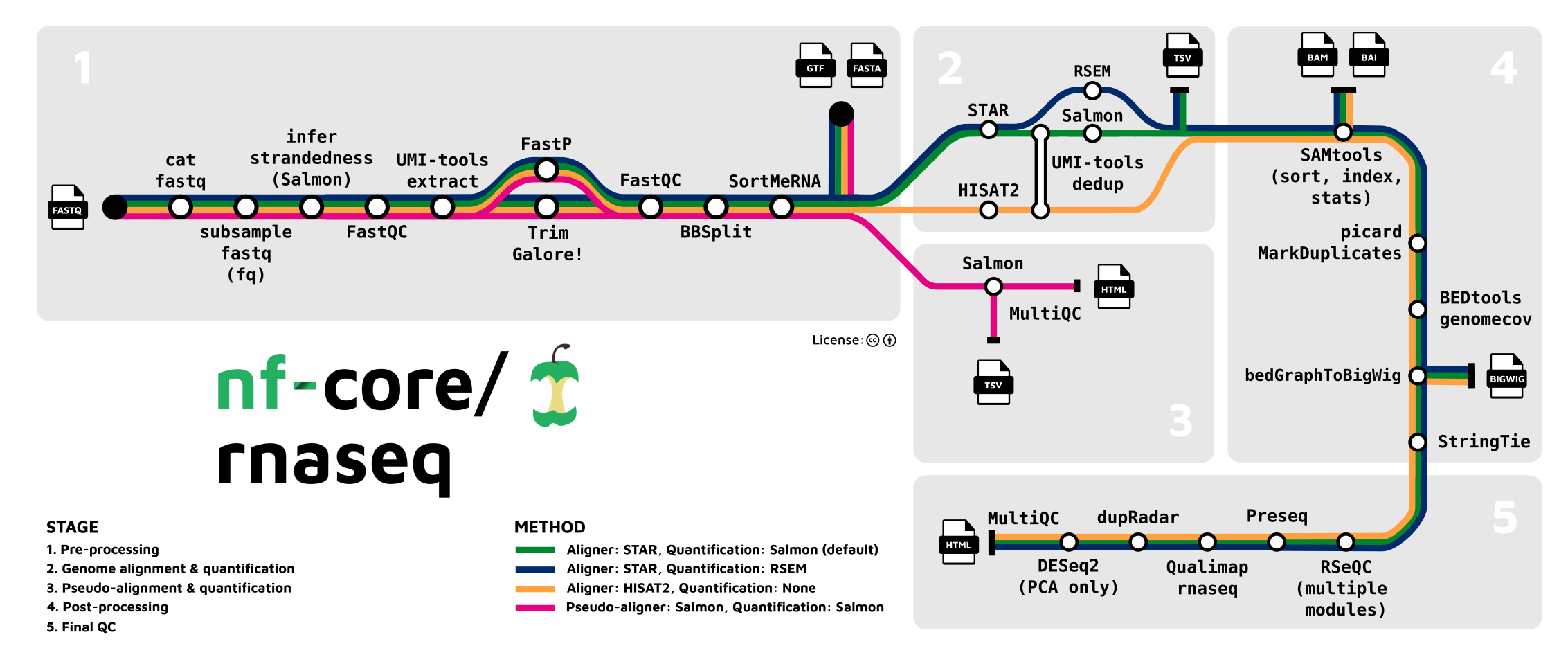
nf-core/rnaseq is a bioinformatics pipeline that can be used to analyse RNA sequencing data obtained from organisms with a reference genome and annotation. It takes a samplesheet and FASTQ files as input, performs quality control (QC), trimming and (pseudo-)alignment, and produces a gene expression matrix and extensive QC report.
Nextflow script to run
The script is called pipe_rnaseq.sh and contains the following lines:
#! /bin/bash
#SBATCH --nodes=1
#SBATCH --cpus-per-task=10
#SBATCH --mem-per-cpu=10G
#SBATCH -t 72:00:00
source /local/env/envnextflow-23.10.0.sh
nextflow run rnaseq \
-profile genouest,singularity \
--fasta /groups/p_aphid/mgalland/data/refs/Pisum_sativum_v1a.fa \
--input samples.csv \
--gtf /scratch/mgalland/Pisum_sativum_v1a_genes.gtf \
--multiqc_title alves \
--trimmer fastp \
--bam_csi_index \
--save_align_intermeds \
--outdir /scratch/mgalland/ \
--skip_rseqc
Run using SLURM + pipe_rnaseq.sh script
# Request resources from genouest
srun -c4 --mem=40G --pty bash
# Launch script on a node with bash
bash pipe_rnaseq.sh
Not done (but good idea for next time)
Specify a version of Nextflow:
-r 1.3.1
Written on January 23, 2024